应用全基因组测序纵深分析中国MRSA
本研究以MRSA克隆复合物(CCS)为主轴,通过全基因组测序延伸分析MRSA的基因背景,纵深挖掘其遗传进化的更替关联。最终确定了中国优势谱系及其相关的基因组和表型特征,强调了在中国监测MRSA的必要性。
MRSA从发现至今感染几乎遍及全球,已成为院内和社区感染的重要病原菌之一。
同济大学附属上海市肺科医院余方友教授课题组的一项研究对中国MRSA的分子流行病学和表型特征进行充分挖掘和探讨,为MRSA流行情况提供了重要线索。该研究发表在《Emerging Microbes & Infections》,2022年影响因子为19.568。
研究策略:
1、全基因组测序及数据分析
从不同年限和医院分离获取的565株MRSA并对其进行全基因组测序后分析MLST分型、spa分型、SCCmec分型、耐药基因和毒力因素等。该研究从克隆分型切入但对细分关联进行了纵向深挖,拔高了文章的可读可研性。
2、表型验证
通过评估MIC水平来检测MRSA对抗菌药物敏感性,验证基因型和表型的关联性。

研究结果:
该研究观察到MRSA克隆复合物谱系主要为:CC59,CC5和CC8。其中CC59和CC8主要是来源血液或渗出液样本而CC5主要来源于痰样本,突出强调不同克隆复合物的耐药表型的差异。
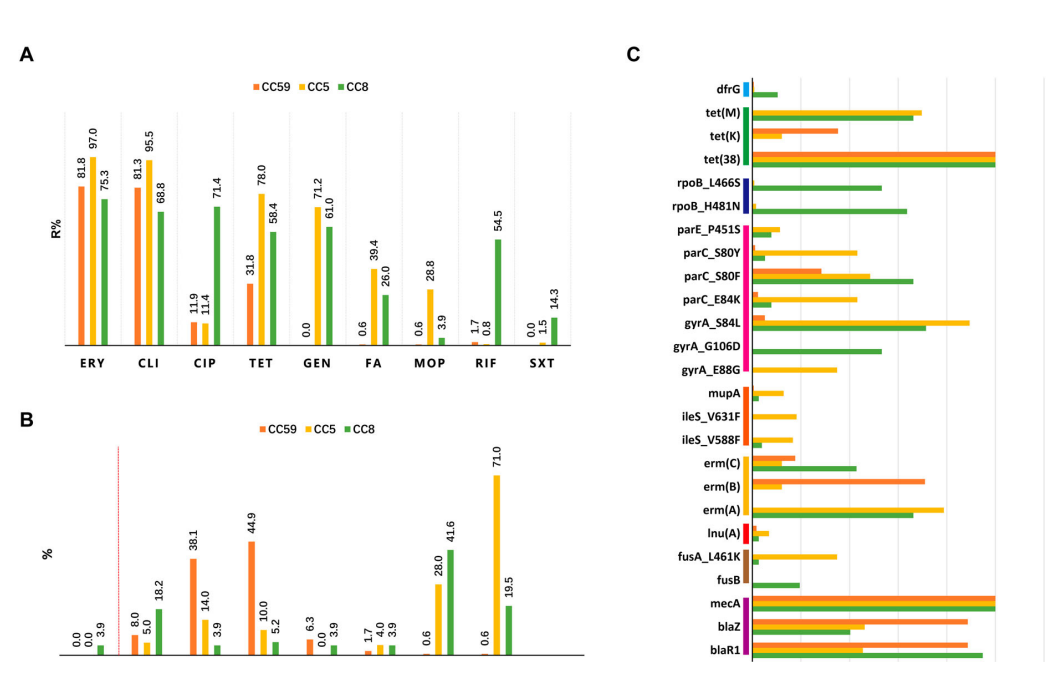
图1. 不同克隆复合物耐药基因型和表型的情况
通过地域分析,发现不同区域流行的MRSA存在的差异。此外,随着时间的推移,发现2014-2015年的主要谱系为CC8,但在2016年后被CC59所取代。CC398主要来源于家畜,最早在欧洲,北美等地方流行,而本研究显示该型别在2014至今在中国一直处于上升趋势。此外,研究观察到CC5群体发展不稳定,其中ST5-t311-II、ST764-t1084-II、ST5-t2460-II和ST764-t002- II存在复杂的竞争。
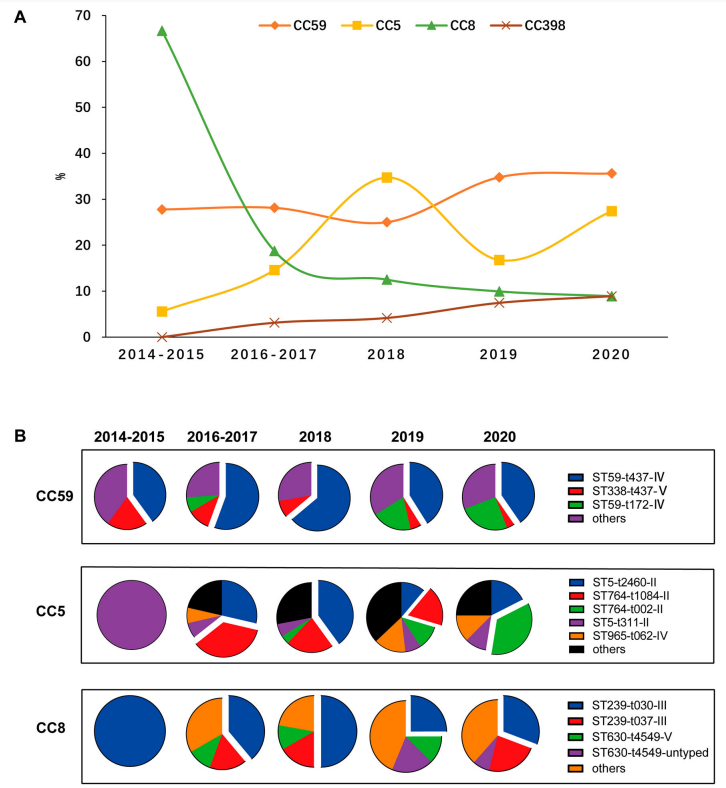
图2. MRSA分离株中发现的主要CCs
进一步与NCBI基因数据库比较发现,中国,欧洲和北美地区的ST59,ST5和ST239分别属于不同的进化分支。其中ST5,在全球大流行中有最多的克隆分支。在进一步的分析表明,毒力决定基因谱和抗生素谱与克隆系密切相关。特别是CC59 MRSA对大多数被测试的抗菌素的耐药性较低,且携带的耐药性决定因子较少,而利福平耐药和莫匹罗辛耐药分别与CC8和CC5关联性更强。该研究特别提出PVL编码基因在ST338、CC30、CC398、ST8和CC22中更为常见;此外tsst-1与ST5相关。
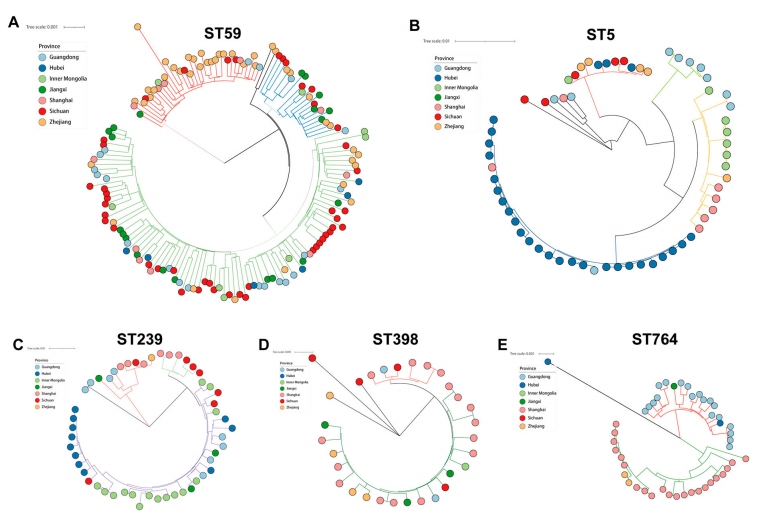
图3. 不同克隆复合物核心基因组系统发育
全文简单的图表清晰的表达了研究结果,通过深挖CCs和表型相关联的基因为MRSA后续风险预测提供参考。
文献来源:https://doi.org/10.1080/22221751.2022.2032373
1、凡本网所有原始/编译文章及图片、图表的版权均属微生物安全与健康网所有,未经授权,禁止转载,如需转载,请联系取得授权后转载。
2、凡本网未注明"信息来源:(微生物安全与健康网)"的信息,均来源于网络,转载的目的在于传递更多的信息,仅供网友学习参考使用并不代表本网同意观点和对真实性负责,著作权及版权归原作者所有,转载无意侵犯版权,如有侵权,请速来函告知,我们将尽快处理。
3、转载请注明:文章转载自www.mbiosh.com
联系方式:020-87680942