iProbiotics--免费快速筛选益生菌的机器学习平台
2021年11月27日,内蒙古农业大学张和平团队和内蒙古大学左永春团队 在Briefings in Bioinformatics(IF:11.622)发表题为 “iProbiotics: a machine learning platform for rapid identification of probiotic properties from whole-genome primary sequences”的研究性论文。
评估新型益生菌菌株的适宜性和潜力是一个复杂的过程,应从多个角度进行细致的评估。目前,验证和选择益生菌最常用的方法是进行随机对照试验 (RCTs)。基本上,在 RCT 中,候选益生菌的临床功效是通过人类干预与一组定义的临床读数参数进行测试的。此外,微生物组研究的新工具得到迅速发展,可以从生理学方面评估益生菌的功能,例如益生菌的肠道定植、物种甚至菌株水平的肠道菌群变化以及施用的益生菌与内源性肠道微生物组/宏基因组之间的相互作用。然而,有报道称临床结果相互矛盾。结果不一致的部分原因是益生菌菌株和配方的不同类型、临床干预方案和个体差异。此外,即使假定益生菌的施用通常是安全的,不会增加健康风险,但临床试验中的安全结果报告不一致,包括对系统性感染的理论风险、易感者的不良代谢和免疫刺激反应、胃肠道副作用和微生物基因转移的描述。因此,解释 RCT 产生的数据仍然存在重大挑战和局限性,它可能并不总是评估益生菌干预有效性和安全性的最合适方法。我们需要更多的研究来收集深入的信息,并从不同的角度发展知识,以确保益生菌在临床上的有效和安全使用。
近年来,学者们已经尝试将机器学习算法应用于比较基因组学挖掘,以识别大规模宏基因组测序数据集中的特定功能特征。目前,美国国家生物技术信息中心(NCBI)数据库(www.ncbi.nlm.nih.gov)包含约6000可公开获得的双歧杆菌和乳杆菌的基因组序列数据集。事实上,大多数特征明确且目前使用的益生菌都属于这两个家族。尽管机器学习已广泛应用于生物学和生物医学领域并显示出广阔的前景,但尚未将机器学习方法应用于构建益生菌预测平台。因此,帮助预测和选择具有所需特征的潜在益生菌菌株以通过实验和 RCT 进一步验证将是有意义的或确实是必要的。因此,我们帮助预测和选择具有理想特征的潜在益生菌菌株,并通过实验和RCT进一步验证,将是有意义且是必要的。
这项工作的主要目的是开发一个基于机器学习算法的平台,从现有的全基因组测序数据中预测益生菌特性。该文构建了三个独立的模型(http://bib.oxfordjournals.org/),分别是模型1:益生菌和非益生菌菌株的预测器;模型2:益生菌乳杆菌、益生菌双歧杆菌和其他益生菌的预测器;模型3:益生菌乳杆菌和非益生菌乳杆菌的预测器。

乳酸菌菌群普遍存在于食品中,其中一些细菌具有益生菌特性。然而,益生菌的发现和实验验证需要大量的时间和精力。因此,开发有效的筛选方法来识别益生菌是非常有意义的。
测序技术的进步产生了大量的基因组数据,使我们能够在这项工作中创建一个基于机器学习的平台来达到这种目的。本研究首先从益生菌数据库(PROBIO)和文献调查中选择了一个全面的益生菌基因组数据集。然后,进行了k-mer(从2到8)组成分析,揭示了菌株基因组中多样化的寡核苷酸组成和益生菌基因组中明显比非益生菌基因组更多的益生菌(P-)特征。为了减少噪音和提高计算效率,通过增量特征选择(IFS)方法对87 376个k-mers进行了细化,模型在184个核心特征时达到最大准确度水平,预测准确率高(97.77%),曲线下面积高(98.00%)。利用基因本体论(GO)、京都基因和基因组百科全书(KEGG)和使用子系统技术的快速注释(RAST)数据库的注释进行功能基因组分析,以及与宿主胃肠道生存/定居、碳水化合物利用、抗药性和毒力因子相关的基因分析,发现P特征的分布偏向与益生菌功能有关的基因/通路。
该研究团队的结果表明,益生菌的作用不是由单一基因决定的,而是由k-mer基因组成分的组合决定的,该文为益生菌的鉴定和潜在机制提供了新的见解。这项工作创造了一个新颖的、免费的在线生物信息学工具iProbiotics,它将促进对益生菌的快速筛选。
图文鉴赏

图1:iProbiotics预测平台的框架。
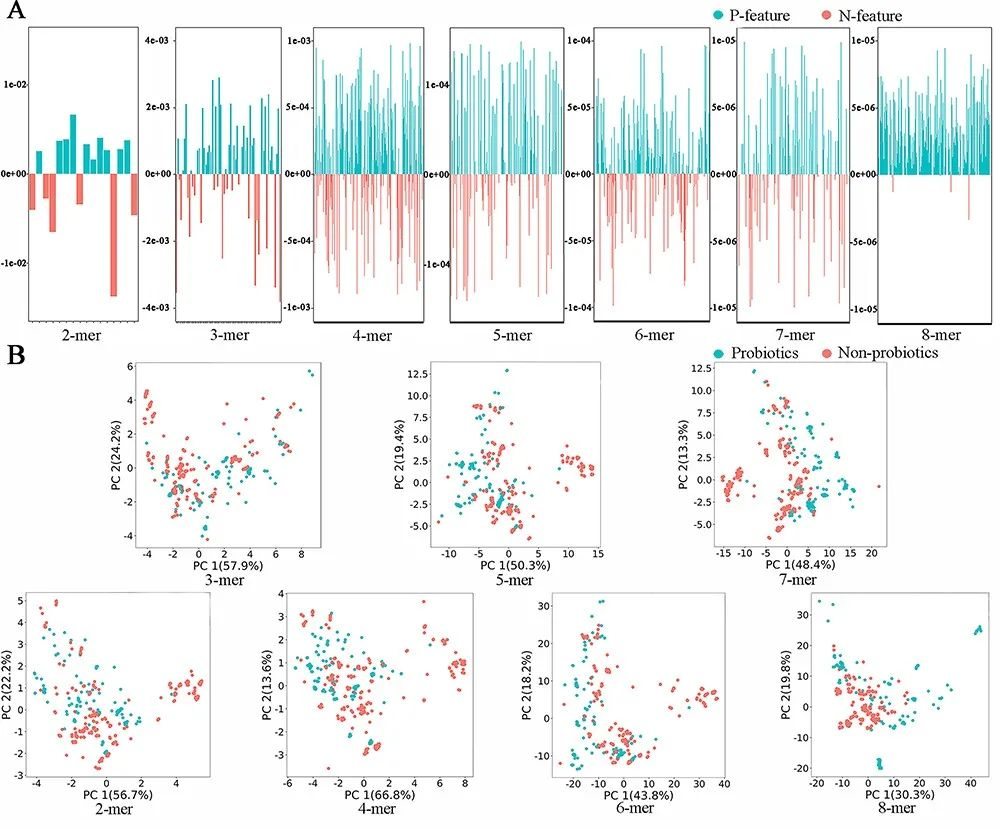
图2:检索到的细菌基因组序列的寡核苷酸组成概况。(A) 通过寡核苷酸组成表示益生菌和非益生菌基因组。y轴是 "Probio651 "所有样品的k-mer值的平均值。益生菌(P-)和非益生菌(N-)特征的不均匀分布显示在每个k-mer组(2-8mer)。(B)不同的k-mer组的益生菌和非益生菌基因组的PCA得分图。图中的每个点代表一个细菌基因组。
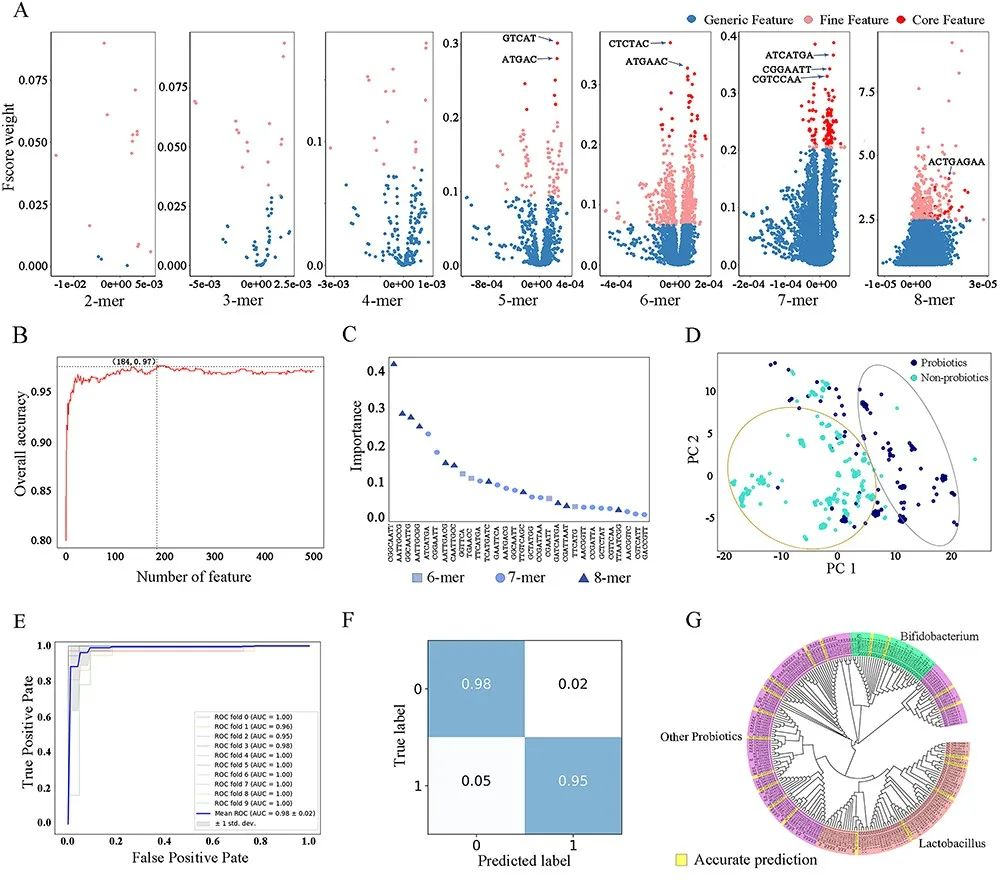
图3:益生菌预测模型的核心特征选择和机器学习训练。(A) 每个k-mer组中一般、精细和核心特征的F-score重要性得分的变化。一般、精细和核心特征是由IFS方法定义的。图中的每个点代表一个特征。(B) 预测精度与核心特征数量之间的关系。184个核心特征实现了最大的预测精度。(C) 核心特征中的前30个(Top 30)特征和它们的重要性。(D) 通过分析益生菌和非益生菌基因组的184个核心特征产生的PCA得分图。图中的每个点代表一个细菌基因组。(E)模型1(益生菌和非益生菌菌株的预测器)的10倍交叉验证ROC曲线。(F)模型1(益生菌和非益生菌菌株的预测器;1:益生菌,0:非益生菌)的混淆矩阵。(G)显示模型2的进化关系的分类树状图。训练组的菌株分别用粉色阴影表示乳杆菌,绿色阴影表示双歧杆菌,紫色阴影表示其他益生菌。准确预测的益生菌以黄色阴影表示。
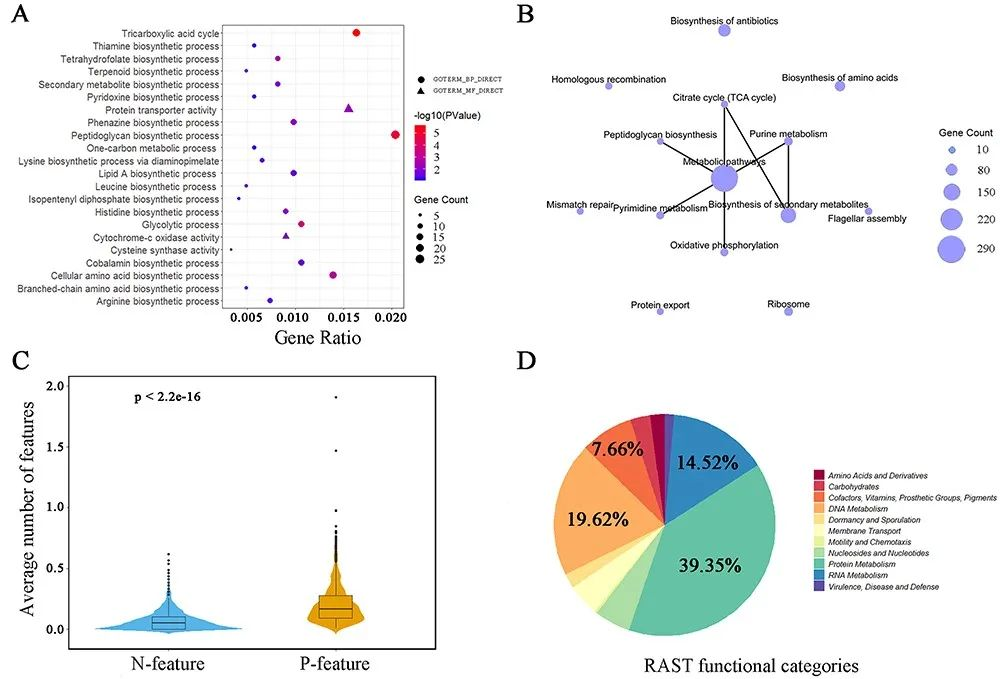
图4:富含核心特征的基因的富集分析。(A) 2282个基因富集在P特征中,这些基因的功能被GO进一步注释。(B) KEGG通路富集分析在这些基因之间产生了一个通路网络。图中的点的大小代表基因的数量,图中的连接代表两个通路之间有共享基因。(C)子系统技术(RAST)揭示了益生菌菌株的核心功能作用,共有3822个序列被分配到60个特定的核心功能作用。箱线图说明了3822个序列中益生菌(P-)和非益生菌(N-)的特征分布。(D) 3822个序列的核心功能作用的分布。
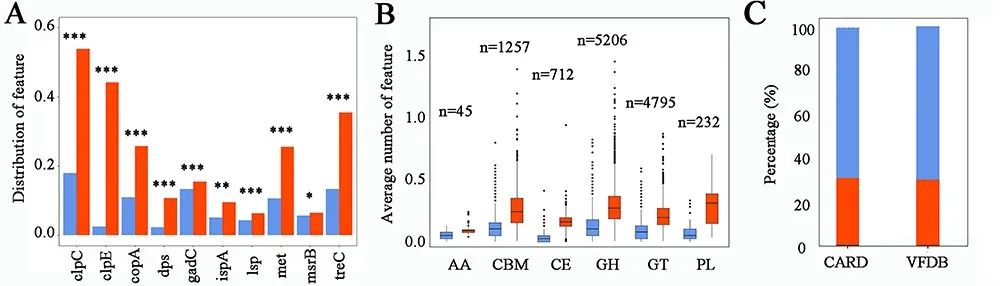
图5:益生菌的核心特征的传统基因组学分析。(A)益生菌(P-,红色)和非益生菌(N-,蓝色)的特征在与宿主胃肠道生存有关的基因序列中的分布。(B) P-特征和N-特征在碳水化合物活性酶编码基因序列(AAs;CBMs;CEs;GHs;GTs和PLs)的分布。(C) 堆积的柱状图显示了P-特征和N-特征(由模型1生成:益生菌和非益生菌菌株的预测器)在2702个抗生素抗性基因(由CARD检索)和3606个毒力因子相关基因(由VFDB检索)中的分布。
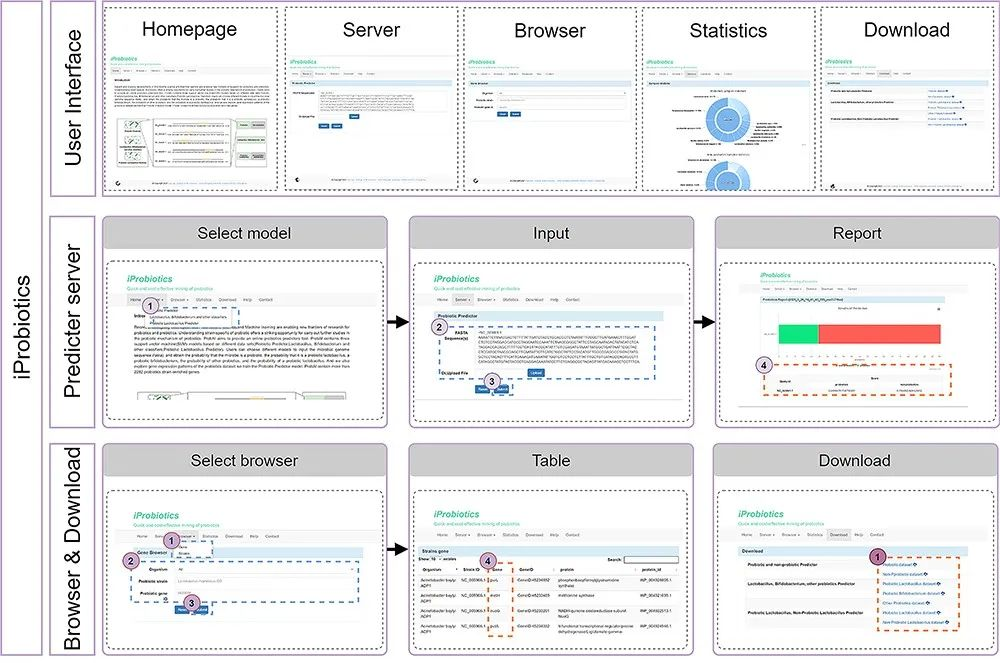
图6:iProbiotics平台的数据可视化和网络服务器使用。
原文链接:https://doi.org/10.1093/bib/bbab477
1、凡本网所有原始/编译文章及图片、图表的版权均属微生物安全与健康网所有,未经授权,禁止转载,如需转载,请联系取得授权后转载。
2、凡本网未注明"信息来源:(微生物安全与健康网)"的信息,均来源于网络,转载的目的在于传递更多的信息,仅供网友学习参考使用并不代表本网同意观点和对真实性负责,著作权及版权归原作者所有,转载无意侵犯版权,如有侵权,请速来函告知,我们将尽快处理。
3、转载请注明:文章转载自www.mbiosh.com
联系方式:020-87680942