
Nature子刊 | smRandom-seq:微生物转录组的单细胞视角


微生物广泛存在于人体和各类环境中,其群落内的转录组异质性对微生物适应环境、耐药性产生及宿主互作等过程至关重要。然而,传统的群体水平转录组学仅能提供平均值,无法揭示单个微生物细胞的基因表达差异。例如,即使在同基因细菌群体中,个体间也可能因环境压力(如抗生素暴露)表现出显著的转录异质性,这种异质性是微生物耐药性和持久性的关键机制。此外,宿主与微生物的相互作用、微生物群落的功能分化(如肠道菌群的代谢分工)等问题,均需在单细胞分辨率下解析。因此,开发单细胞层面的微生物转录组测序技术成为突破现有研究瓶颈的关键。近年来,单细胞RNA测序(scRNA-seq)技术在真核生物研究中取得了巨大进展。然而,微生物单细胞RNA测序面临多重挑战:mRNA缺乏poly(A)尾使传统poly(dT)引物失效,RNA含量比哺乳动物细胞低约2个数量级且rRNA占比超80%;细胞壁结构复杂导致裂解困难;以及现有方法多局限于实验室菌株。
基于此,浙江大学王永成团队开发了smRandom-seq高通量单微生物RNA测序技术(实验流程见图1-图4,具体操作请见原文),通过三大创新突破瓶颈:①双重条形码标记:12个独立反应管的预索引随机引物与液滴内条形码珠(含poly(dT)-UMI-条形码)结合,实现“管内预索引+液滴特异性标签”双重标记,确保单细胞精准追溯;②CRISPR-Cas9靶向耗竭rRNA:针对实验室培养菌株(如大肠杆菌、金黄色葡萄球菌),设计sgRNA池切割rRNA-derived cDNA,将rRNA污染从70%–90%降至10%–50%;③定制化样本处理:革兰氏阴性菌用溶菌酶、阳性菌及复杂样本用溶菌酶+溶葡萄球菌素酶解,37℃ 15分钟高效破壁。
凭借这些创新,该技术在多领域展现潜力:①耐药性研究:分析数千个单个大肠杆菌细胞在抗生素处理下的转录组变化,成功鉴定出具有独特基因表达模式的抗生素抗性亚群;②复杂群落解析:在人类肠道菌群中揭示拟杆菌属与crAss样噬菌体的协同进化关系,在牛瘤胃中鉴定172个核心物种及12个碳水化合物代谢功能簇;③方法学革新:建立标准化流程并开源代码(GitHub),降低技术门槛。未来,smRandom-seq有望与空间转录组、单细胞蛋白组等多组学技术整合,推动精准医疗、工业微生物筛选及生态监测等领域的发展。
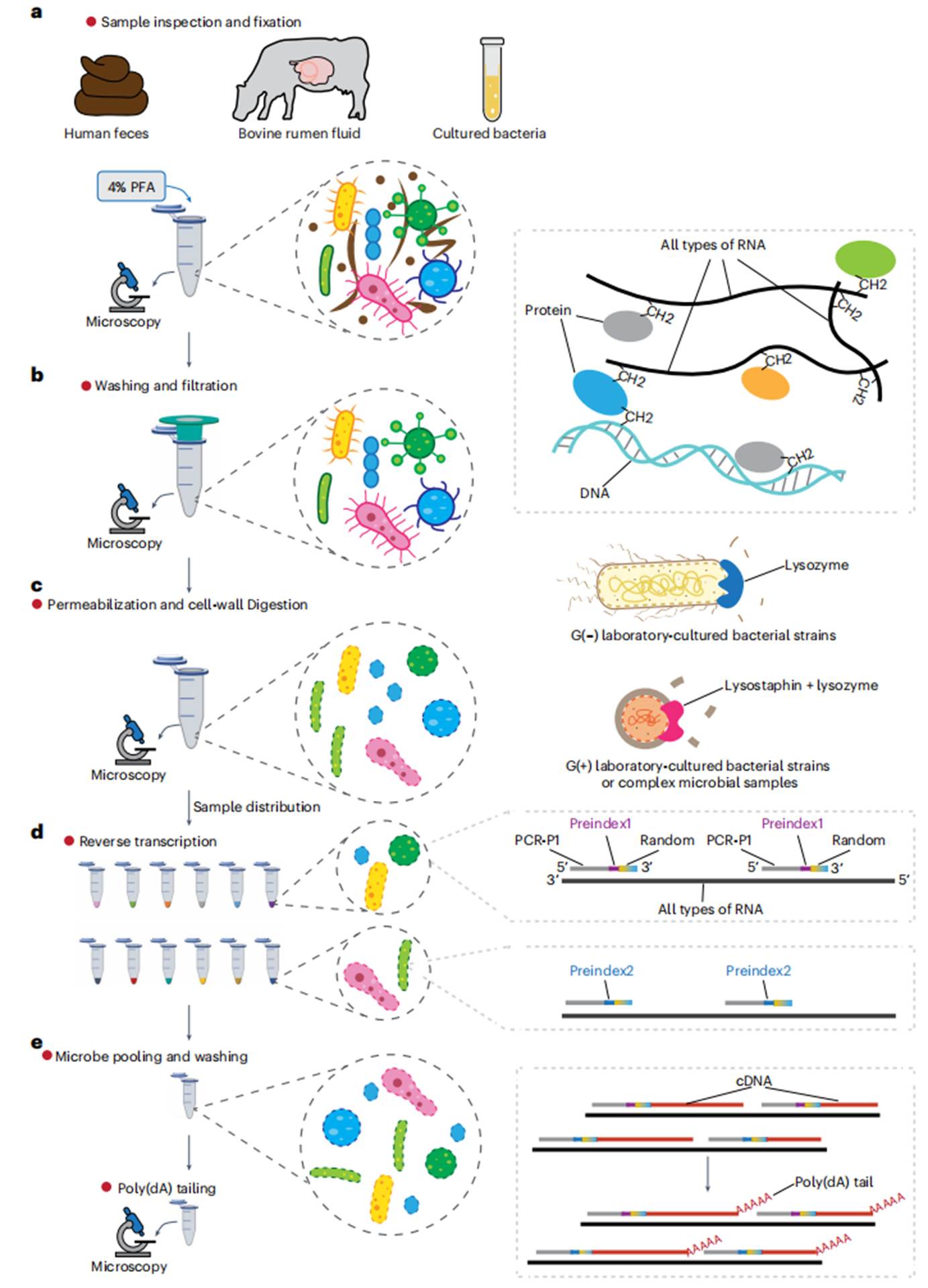
图1 样本处理和原位反应的工作流程。(a)使用冰浴的4%多聚甲醛(PFA)固定微生物样本(8–18小时),通过交联RNA、DNA和蛋白质维持转录组完整性。不同样本(如人类粪便、牛瘤胃液)需按规范操作,避免RNA降解。(b)通过40μm→10μm细胞滤网过滤,去除宿主细胞、食物残渣等大颗粒杂质,确保获得纯净的单细胞悬浮液(杂质占比需<5%)。(c)用含0.04%吐温-20的Perm缓冲液透化细胞膜(冰上孵育3分钟)。革兰氏阴性菌(如大肠杆菌)仅用溶菌酶(2.5μg/μL);革兰氏阳性菌(如金黄色葡萄球菌)或复杂样本(如人类粪便)需联合溶菌酶(2.5μg/μL)+溶葡萄球菌素(12.5ng/μL),37°C酶解15分钟。(d-e)原位逆转录(RT)和poly(dA)尾添加:在12个独立的PCR管中进行原位逆转录反应,每个管中加入带有独特预索引序列的随机引物,将RNA转录为预索引cDNA,并在cDNA的3'端添加poly(dA)尾。离心洗涤去除未结合引物(保留>40%微生物细胞)。
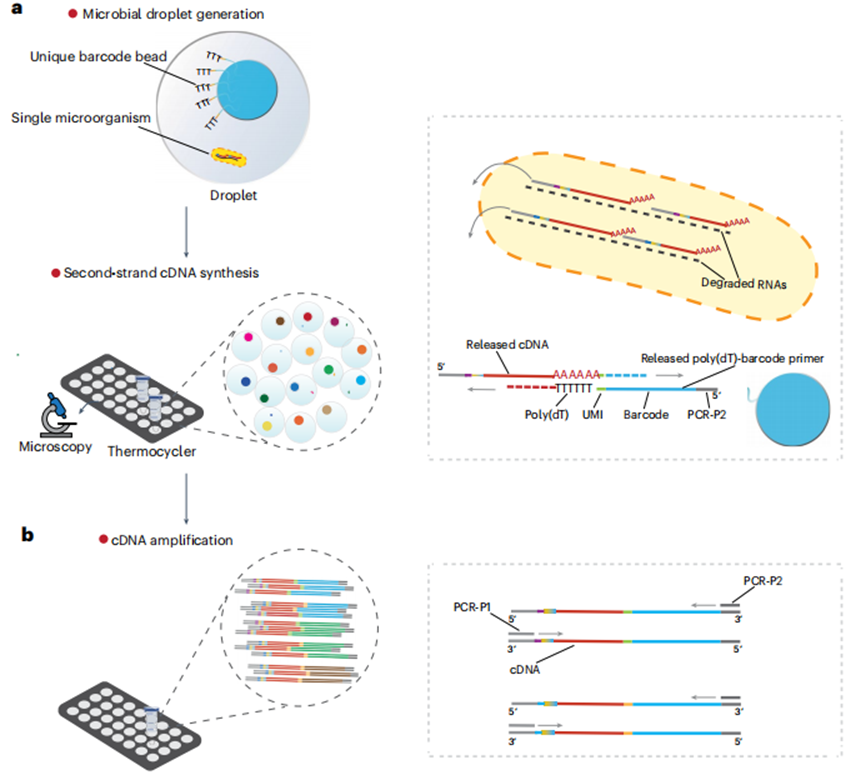
图2 单微生物条形码文库构建的工作流程。(a)使用商业化设备(如M20 Genomics)或自制平台,将微生物悬液(调整至2,000细胞/μL)、条形码珠(含poly(dT)-UMI-条形码-PCR引物柄)和反应试剂封装为~100μm液滴,每个液滴含单个细胞和一个条形码珠。条形码珠释放poly(dT)引物,通过与cDNA的poly(dA)尾结合,标记细胞特异性条形码和UMI。在条形码珠子上的poly(dT)序列与cDNA的poly(dA)尾结合后,使用DNA聚合酶合成第二链cDNA。(b)PCR扩增:破碎液滴后,使用特异性的PCR引物(PCR-P1和PCR-P2)进行PCR扩增,目标片段大小为100–1,000 bp,<100 bp片段需<20%。
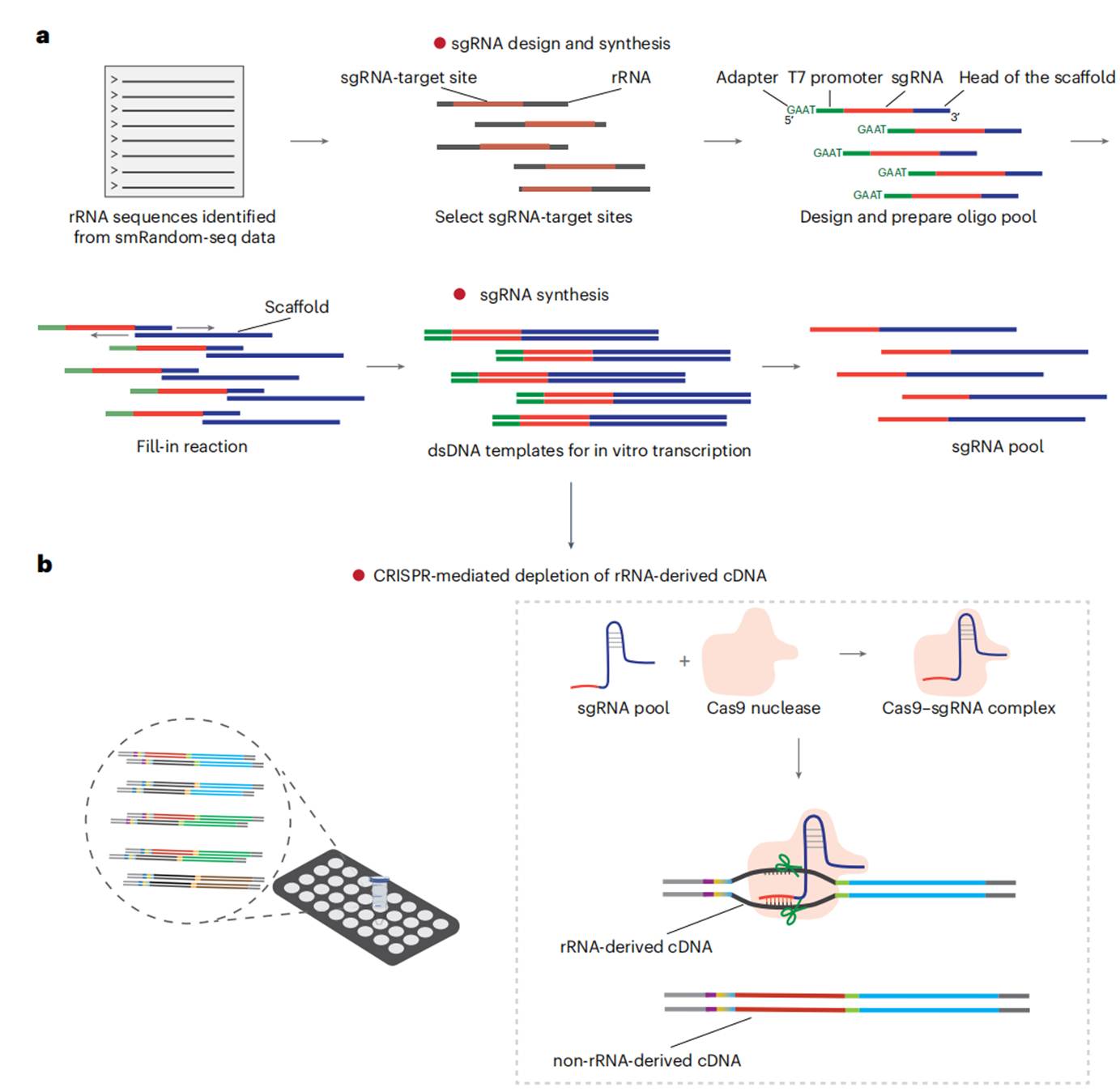
图3 文库富集的工作流程。(a)sgRNA设计和合成:基于smRandom-seq数据识别目标rRNA序列,用DASHit 2.0软件设计sgRNA(每50 bp选取一个靶点,GC含量5–15%),合成含T7启动子的sgRNA前体,经体外转录获得sgRNA池。(b)CRISPR-Cas9介导的rRNA耗竭:将Cas9-sgRNA复合物与cDNA文库混合(cDNA:Cas9:sgRNA = 1:100:1,000),37°C孵育2–4小时,特异性切割rRNA-derived cDNA,使rRNA占比从70–90%降至10–50%。注:人类肠道、牛瘤胃等样本因rRNA多样性高,不适用酶法耗竭,需通过增加测序深度(~100 Gb/样本)覆盖转录组。
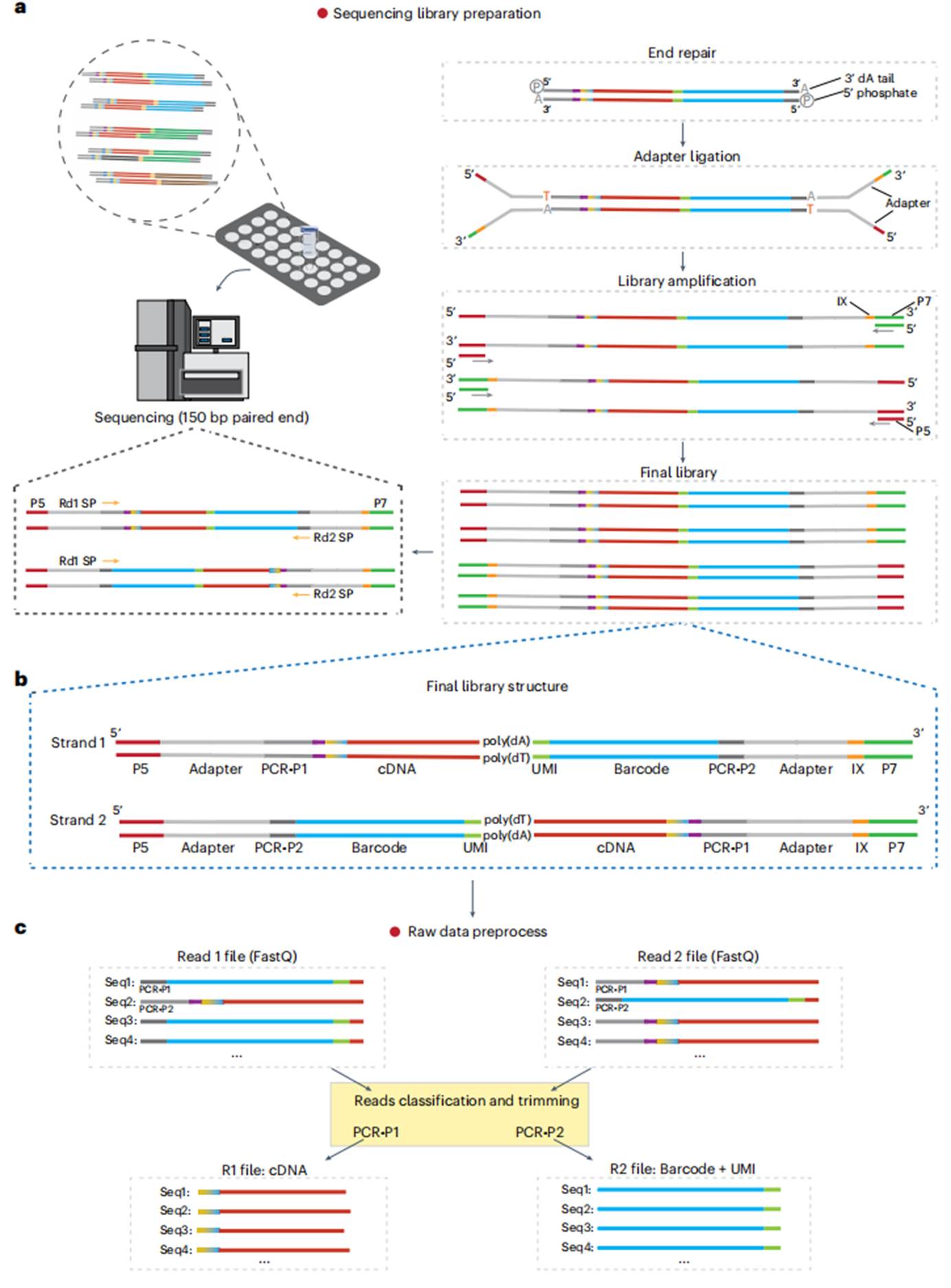
图4 测序文库准备和原始数据预处理。(a)测序文库准备:对经过rRNA耗竭和PCR扩增后的cDNA文库进行进一步处理,使其适合进行高通量测序。进行末端修复、接头连接、文库扩增和纯化,确保文库的片段大小(300-800 bp)和浓度符合测序平台的要求。(b-c)原始数据预处理:对测序生成的原始数据(FASTQ文件)进行预处理,用cutadapt等工具去除接头序列,根据PCR-P1/PCR-P2标签区分链(R1含cDNA序列,R2含条形码+UMI),过滤低质量reads。生成高质量的数据文件,包含每个单细胞的基因表达信息。
1、凡本网所有原始/编译文章及图片、图表的版权均属微生物安全与健康网所有,未经授权,禁止转载,如需转载,请联系取得授权后转载。
2、凡本网未注明"信息来源:(微生物安全与健康网)"的信息,均来源于网络,转载的目的在于传递更多的信息,仅供网友学习参考使用并不代表本网同意观点和对真实性负责,著作权及版权归原作者所有,转载无意侵犯版权,如有侵权,请速来函告知,我们将尽快处理。
3、转载请注明:文章转载自www.mbiosh.com
联系方式:020-87680942

